

Review on Chemistry of Natural and Synthetic Indolizines with their Chemical and Pharmacological Properties
- *Corresponding Author:
- Dr. Katharigatta N. Venugopala
Department of Pharmaceutical Sciences, College of Clinical Pharmacy, King Faisal University, Al-Ahsa 31982, Kingdom of Saudi Arabia.
E-mail: kvenugopala@kfu.edu.sa
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Abstract
This review emphasizes chemistry of synthetic indolizine analogues including chemical reactions in addition to natural indolizidine alkaloids and their physical and pharmacological properties. Synthetic indolizine analogues for various pharmacological properties such as central nervous system depressant, analgesic and anti-inflammatory, anticancer, antibacterial, antioxidant, and larvicidal and anti-HIV are reported along with their chemical reactions
Keywords
Analgesic and anti?inflammatory, antibacterial, anticancer,- antioxidant, central nervous system depressant, indolizidine alkaloids,- indolizine, larvicidal and anti?HIV
Introduction
The history of indolizine goes back to 1890 when the Italian chemist- Angeli [1] reported the preparation of the imine?anhydride (1) of- pyrroylpyruvic acid and suggested the name pyrindole for the completely- unsaturated parent base (2). Twenty?two years later, in 1912, Scholtz [1]- reported the first synthesis of compound (2). He treated 2?methylpyridine- with acetic anhydride at 200–220°C to give what he called “picolide,” acid- hydrolysis of which afforded a colorless crystalline solid (subsequently- identified as compound [2]) which had weakly basic properties. In view- of this observation, it was speculated that this compound could not- be a true derivative of pyridine. Furthermore, this new compound- gave reactions characteristic of pyrroles and indoles and had the same- empirical formula (C8H7N) as indole and isoindole. In light of these- observations, Scholtz ascribed the pyrrolopyridine structure (2) to his- new compound and named it pyrrocoline but later adopted the name- pyrindole as suggested by Angeli.[1] The validity of Scholtz’s formulation- was confirmed by Diels and Alder,[1] who established the presence of four- double bonds by catalytic reduction of pyrrocoline to a derivative (3),- which on treatment with cyanogen bromide gave a product shown to- be identical in all aspects with (±) coniine (4) [Figure 1] previously- prepared by Loffler et al. At present, the compound previously known- as pyrrocoline or pyrindole is known as indolizine with the following- numbering (2a).

Figure 1: Reagents (i) cyanogen bromide
Indolizidines as natural products
Indolizidines [Figure 2] are commonly distributed in nature, especially- in plants. Their structures can be described either as analogs of the- aromatic bicyclic indolizine or as azabicyclo[4.3.0]nonanes.[2]

Figure 2: The bicyclic core of indolizidine alkaloids (2b)
The indolizidine alkaloids exhibit a wide range of pharmacological- properties [3,4] and have been the focus of various synthetic- studies.[5?15] Most of the naturally occurring indolizidines have- been isolated from species of the genus Dendrobates (poison?arrow- frogs), Dendrobium (orchids), Leguminosae family (plants), Monomorium (ants), and Tylophora. Among them, the lipophilic- pumiliotoxins and hydrophilic polyhydroxy indolizidines are the two- most important classes of compounds.
Pumiliotoxins
A large variety of structurally distinctive and pharmacologically- active compounds are isolated from amphibians. The distinctive- source is the skin secretions of certain brightly colored frogs native- to the rain forests of Western Colombia and Panama. Likely, since- pre?Columbian times, these frogs have been employed by the- Noanama and Embera Indians to prepare poison blow darts.[16,17] The- first description of darts envenomed with skin secretions of poison- frogs dated from 1825 and described the use of a single frog to charge- at least twenty blow darts. The chemistry and pharmacology of- “poison dart” frogs of the family Dendrobatidae were pioneered by- Witkop, Daly et al.[18,19] To date, more than 300 organic compounds- have been isolated from this amphibian family, the vast majority of- which are unique to dendrobatid frogs.[20] The pumiliotoxin A and- allopumiliotoxin classes of dendrobatid alkaloids are a group of ~40- alkylidene indolizidine alkaloids that display particularly significant- pharmacological activities.[21,22] Pumiliotoxins A and B were the- second and third dendrobatid alkaloids to be isolated and were initially obtained in 1967 from skin of the Panamanian poison frog- Dendrobates pumilio.[23] Elucidation of the structure of these alkaloids- was complicated by their instability in acid, likely due to their allylic- hydroxyl group. The structure of these toxins remained unknown until- 1980 when a simpler alkaloid, pumiliotoxin 251D, was isolated as the- major alkaloid component of skin extracts of the ecuadorian poison- frog, Dendrobates tricolor (Epipedobates tricolor). Single?crystal X?ray- analysis of pumiliotoxin 251D hydrochloride finally provided the key- for revealing the constitution of the pumiliotoxin A alkaloids.[24]
The pumiliotoxin alkaloids are characterized by the bicyclic- 8?hydroxy?8?methyl?6?alkylidene?indolizines ring system [Figure 3].- The allopumiliotoxins contains an additional hydroxy group at C?7.- Besides these two main classes, another bicyclic alkaloid, namely,- homopumiliotoxin 223G, has been isolated in small quantities from- the Panamanian poison frog D. pumilio and its structure analyzed.[25?27]- In this compound, the indolizines moiety is replaced by quinilidizine- ring.

Figure 3: Representative pumiliotoxin and allopumiliotoxin alkaloids.
Due to the great number of pumiliotoxins, only a few have been given- a common name. Pumiliotoxins A and B are relatively toxic and a- subcutaneous dose of pumiliotoxin B of 20 μg can cause death in mice.- Recent studies show that pumiliotoxin B binds to an unique modulatory- site on the voltage?dependent sodium channel and enhances sodium- influx.[28,29] This ion flow stimulates phosphoinositide breakdown, which- is believed to be ultimately expressed as cardiotonic and myotonic- activities. Structure–activity studies of natural alkaloids and synthetic- analogs have shown that the structure of the side chain is critical for- these pharmacological activities.[28?33] The intriguing pharmacological- properties and the low availability of the compounds from their natural- sources have led to the development of numerous syntheses of the- pumiliotoxins. Important synthetic contributions have been made by- the groups of Franklin and Overman [34] and Trost and Scanlan,[35] which- have synthesized key members of the class of pumiliotoxins. When- contemplating the design of a total synthesis strategy for the pumiliotoxin alkaloids, attention is immediately drawn to the (Z)?alkylidene side chain.- This unit presents particular problems since stereocontrolled synthesis- of exocyclic alkenes is difficult to achieve. The prospects for the selective- generation of the (Z)?side chain by a Wittig type functionalization of a- suitable precursor indolizidine ketone are low. Therefore, the majority- of synthetic approaches have focused on the selective generation of- an acyclic configurational stable alkene fragment that was used in a- cyclization to generate the pumiliotoxin framework.
Chemistry of Indolizines
Generally, there are three major approaches to indolizine synthesis,- viz., (1) condensation reactions; (2) 1,3?dipolar cycloadditions; and (3)- 1,5?dipolar cycloadditions.
Synthesis of indolizines by condensation reactions- Synthesis of indolizines by reactions of 2?methylpyridine and its- derivatives with acetic anhydride (Scholtz’s reaction)
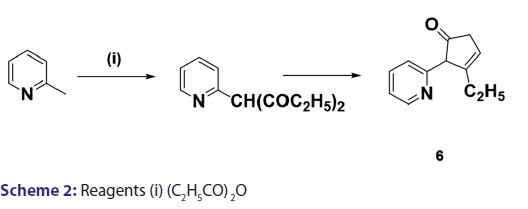
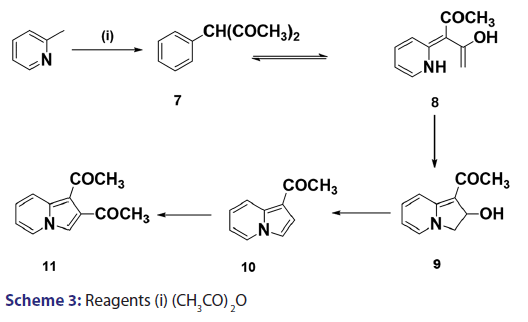
Scholtz’s synthesis [1] of the first indolizine simply involved treating- 2?methylpyridine with acetic anhydride at very high temperature to- yield a crystalline compound, which he called “picolide” and which,- on hydrolysis, afforded the indolizine (2) [Scheme 1]. The principal- difficulty Scholtz faced was in elucidating the structure of “picolide”- and in rationalizing its formation. The presence of one carbonyl- group in “picolide” was clearly established by the formation of oxime,- hydrazone, and semicarbazone derivatives. However, it did not give- any reaction typical of aldehydes. Furthermore, “picolide” was found- to possess only feeble basic properties. Taking into account the above- observations, Scholtz and Fraude [1] speculated that the nitrogen was- acylated and proposed the most suitable structure for “picolide” to- be 1?acetyl?2?methyl?4?ketopyrridocoline (5). If “picolide” was to- have the above?mentioned structure, it would be necessary for it to- undergo a complicated initial cleavage, followed by ring closure to a- five?membered pyrrole in order to explain satisfactorily the formation- of indolizine (2) upon its hydrolysis. Moreover, this structure did not- explain the observed formation of “picolide” from the reaction of one- mole of propionic anhydride with 2?methylpyridine, and consequently,- it was suggested that “picolide” be assigned structure (6) [Scheme 2].- However, Tshitschibabin and Stepanow [1] doubted the validity of Scholtz’s- conclusions and, in 1929, they reinvestigated the synthesis and- formulated “picolide” as 1,3?diacetylindolizine (11) [Scheme 3]; since- 1929, the Scholtz reaction has gained widespread popularity and has- been adopted as a general route to indolizines.
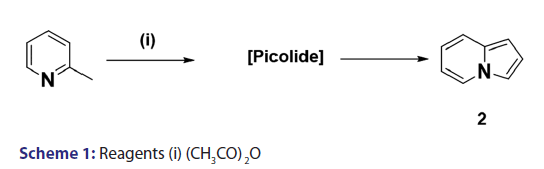
Synthesis of indolizines by ring closure of the pyridinium- salts (Tshitschibabin reaction)
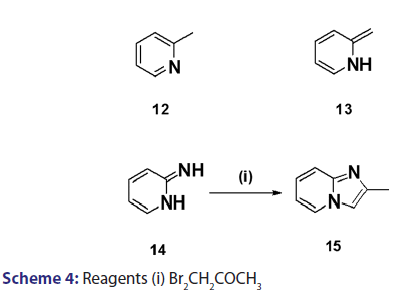
A novel approach to the synthesis of 2?substituted indolizines was- developed, in 1927, by Tschitschibabin.[36] They speculated the existence- of tautomerism in α and β?alkylated pyridines and suggested that- if, for instance, 2?methylpyridine (12) and an α?halogenoketone- were reacted in the presence of an alkali, 2?methylpyridine could- react as the aromatic system (12) or as its tautomer (13), reaction in- the latter form (13) would then present good possibilities for ring- closure, analogous to that observed for compound (14) [Scheme 4].- Speculative as this was, it led to the successful synthesis of 2?substituted- indolizines (17) through cyclization of quaternary pyridinium- salts (16) [Scheme 5]. This approach was fully exploited and several- 2?alkyl? and 2?aryl?indolizines were synthesized. Krohnke el al. [1]- demonstrated that quaternary compounds would react with bases of- suitable strength to afford “enol?betaines” which then undergo “acid- cleavage” losing an acyl group. In view of this observation, these authors- suggested formation of an “enol?betaines” as an intermediate in the Tshitschibabin reaction, ring closure of which would afford an indolizine- [Scheme 5]. However, before long, certain drawbacks become apparent.- When α?halogenated aldehydes were used, the quaternary salts were- not very easily formed and did not always cyclize to form the required- indolizines. In 1965, Hurst et al.[37] observed that the Tshitschibabin- reaction of ethyl 2?quinolylacetate with phenacyl bromide afforded- 2?ethoxycarbonylmethylene?1?phenacyl?1,2?dihydroquinoline (18)- which, upon treatment with sodium bicarbonate, did not undergo ring- closure as expected. Nevertheless, when treated with boiling acetic- anhydride, compound (18) underwent an intramolecular aldol?type- condensation to afford the benzindolizine (19).[38]
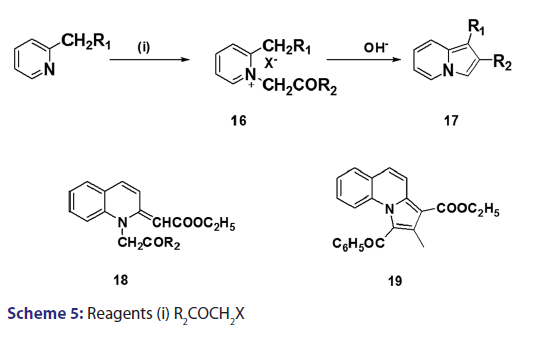
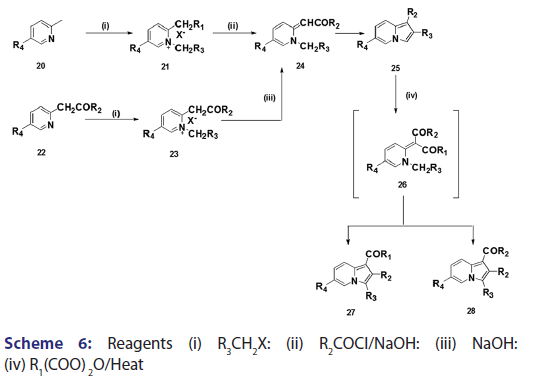
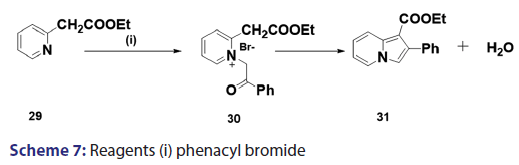
This modified Tshitschibabin indolizine synthesis was successfully- extended to the synthesis of the indolizines (25), (27), and (28) from- the intramolecular aldol?type condensation of the corresponding- acyl methines (24) [Scheme 6].[39] In 1946, Borrows et al.[40,41]- attempted the synthesis of acyl and alkoxy carbonyl indolizines from- α?halogeno?β?diketones and α?halogeno?β?ketoesters using- Tshitschibabin method. However, they could not form the quaternary salt,- and the attempt failed almost two decades later, Bragg and Wibberley [42]- successfully synthesized ethyl 3?acetyl?2?methylindolizine?l?carboxylate- and diethyl 2?methylindolizine?l, 3?dicarboxylate from ethyl- chloroacetate and 3?chloro?pentane?2,4?dione, using a method which- did not require isolation of the quaternary salt. Bragg and Wibberley- also synthesized alkyl and aryl indolizine?l?carboxylates from- α?halogenoketones and ethyl 2?pyridylacetate.[42] When they treated phenacyl bromide with ethyl 2?pyridylacetate (29), ethyl 2?pyridylacetate- hydrogen bromide (30), instead of the quaternary salt was formed from- which the ethyl?2?phenylindolizine (31) crystallized [Scheme 7]. From- the above results, the authors suggested that a part of the ester was- behaving as a base, removing hydrogen bromide and leading to ring- closure. This approach opened new doors to indolizine synthesis.
Synthesis of indolizines by ring closure of- 3?(2?pyridyl)?1?propanols and their analogs
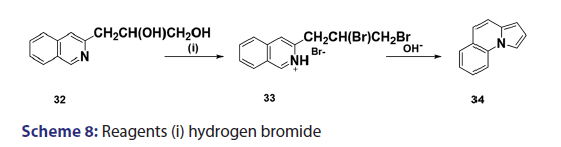
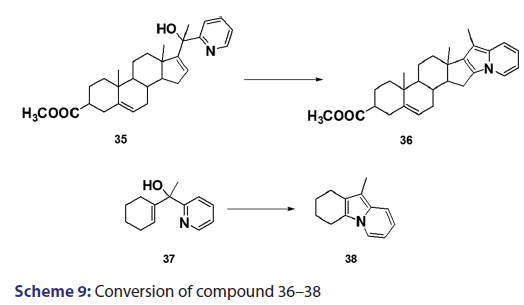
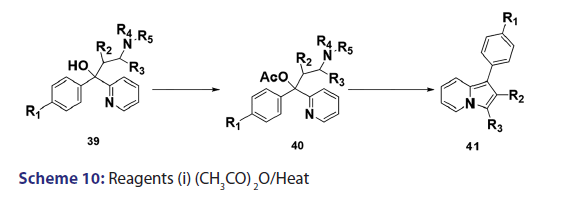
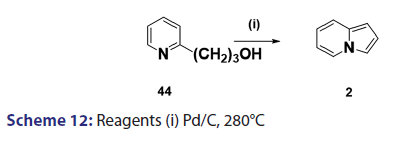
In 1955, Roberts et al.[43] treated 3?(2?quinolyl)?1,2?propanediol (32)- with hydrobromic acid and subjected the product (33) to steam- distillation from alkali to afford 5,6?benzindolizine (34) in very- high yield [Scheme 8]. This remarkable success caught the interest of- chemists worldwide. A year later, two German chemists [37] successfully- extended this novel approach to the synthesis of the highly substituted- indolizines (37) and (38) from the unsaturated alcohols (35)- and (36) [Scheme 9]. During the next 2 years, Barrett and Chambers [44]- reinvestigated Boekelheide’s method and conclusively established- the ability of amino groups to act as good leaving groups. When they- refluxed 3?amino?1?aryl?1?(2?pyridyl)?alkan?1?ols (39) with acetic- anhydride, compounds (40) formed, which cyclized with the elimination- of the amino and acetoxy groups to afford the l?aryl indolizines (41) [Scheme 10]. This approach was used to synthesize several indolizines- including some aza indolizines in high yield.[45,46] Before 1957, many- chemists such as Scholtz,[1] Borrows and Holland,[47] and Diels and Alder,- to mention a few, had reported the synthesis of the parent indolizine (2)- but never in satisfactory yield. Boekelheide and Windgassen- subsequently synthesized the parent indolizine with an overall yield- of 35% from 2?(3?hydroxypropyl)?pyridine?n?oxide (42) [Scheme 11],- not long after, Boekelheide and Windgassen, bettered this yield.[48] They- found that pyrolysis of easily available 3?(2?pyridyl)?l?propanol (44) at- 280°C in the presence of palladium?carbon afforded indolizine (2) in- 50% yield [Scheme 12].
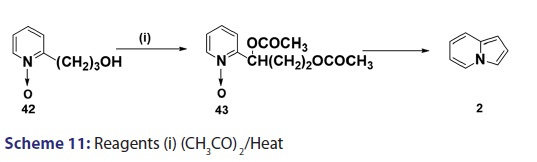
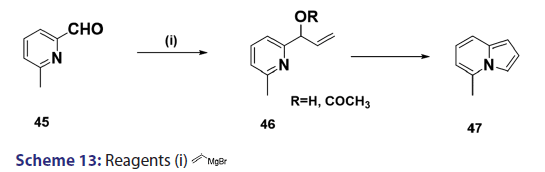
Boekelheide and Windgassen [48] also developed methods for- synthesizing indolizines with no substituents on the five?membered ring.- For example, they treated 6?methylpyridine?2?carboxaldehyde (45)- with vinyl magnesium bromide to give (in 68% yield) the vinyl- alcohol (46, R = H), which was acetylated and the resulting acetate- was then subjected to pyrolysis at 450°C to afford the five?methyl- indolizine (45) [Scheme 13].
Synthesis of indolizines by condensation reactions of heterocyclic- nitrogen compounds with acetylenic and olefinic compounds
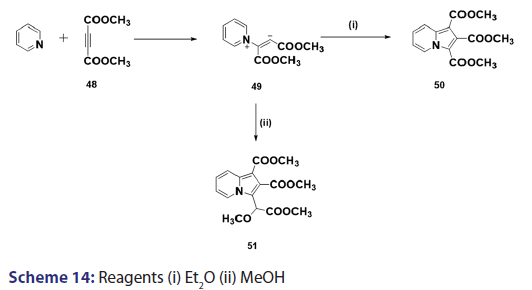
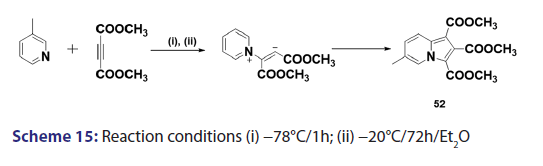
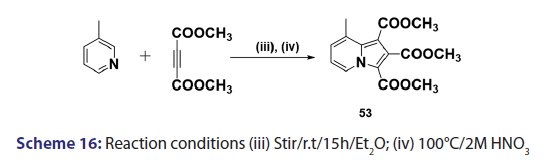
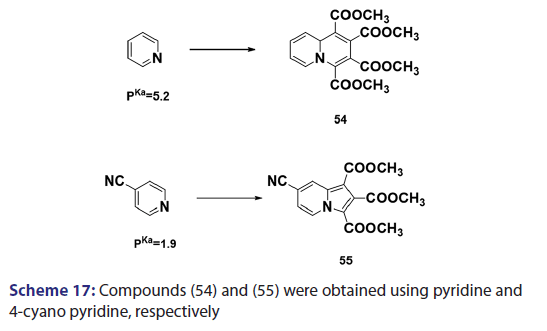
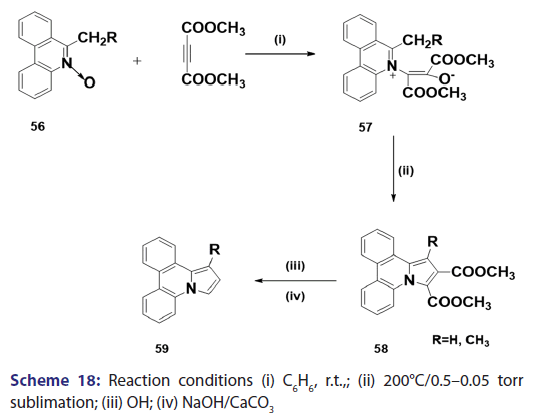
This approach was first introduced by Diels et al. in 1932. They isolated- indolizines (50) and (51) from the intermolecular condensation- of pyridine with acetylene dicarboxylate (48) [Scheme 14]. This approach used the interest of Wiley and Knabeschuh,[49] and, in 1953,- they synthesized the indolizine derivative (52) in 29% yield, from- 3?methylpyridine and acetylene dicarboxylate [Scheme 15]. Seven- years later, Acheson and Plunkett[50] tried the same reaction under- different conditions, without much success. They only managed to- synthesize the indolizine (53) in 6% yield [Scheme 16]. Of particular- relevance is a report published in 1968 by Acheson and Robinson, [51] in- which they related the basicities of pyridines to their reactivity toward- dimethyl acetylene dicarboxylate. It was found that when pyridine- (pKa value 5.2) was used in the reaction, a quinolizine derivative (54)- was formed [Scheme 17]. However, when 4?cyano pyridine was- used which has a much lower pKa value (1.90), trimethyl 7?cyano?l,- 2,3?indolizine tricarboxylate (55) was synthesized [Scheme 17]. When- pyridines with pKa values lower than 1.45 were used, no reaction took- place at all. In 1966, Acheson et al.[52] synthesized dimethyl dibenzo- indolizine?2,3?dicarboxylates (58) from the condensation reaction of- phenanthridine 5?oxide (56) and dimethyl acetylenedicarboxylate.- Compound (57) was the intermediate which, on sublimation, cyclized- to (58) from which the first parent heterocycle (59) (R = H) was- prepared, although derivatives of this novel heterocycle were known- from 1962 [Scheme 18].
Synthesis of indolizines using miscellaneous condensation- reactions
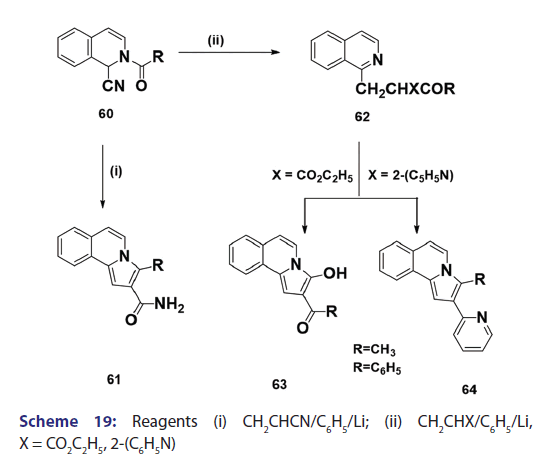
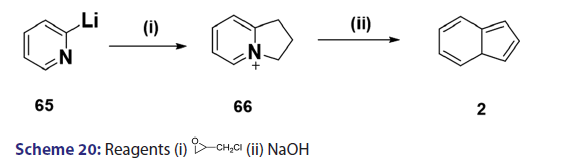
Michael condensation of compounds of the type (60) with- αβ?unsaturated compounds provides another general route to indolizine synthesis. This method was introduced as early as 1953 by Boekelheide- and Godfrey.[53] The authors observed that when acrylonitrile was used,- the corresponding 3?substituted 2?amino carbonyl benzindolizine (61)- was formed [Scheme 19]. On the other hand, when 2?vinylpyridine or- ethyl acrylate was used, ketones of the type (62) were formed which,- on heating or treatment with 100% phosphoric acid, underwent ring- closure to afford the corresponding benzindolizines (63) and (64),- respectively [Scheme 19]. Of particular interest was a paper published in- 1969 by two German chemists.[37] They reported one of the most powerful- methods for the synthesis of parent indolizine (2). Condensation- reaction of 2?pyridyllithium (65) with 2?chloromethyloxirane afforded- first the 2?hydroxy?2,3?dihydro?1H?indoliziniumchloride (66)- which, on treatment with 30% sodium hydroxide, gave the parent- indolizine (2) [Scheme 20].
Synthesis of indolizines by 1,3?dipolar cycloaddition
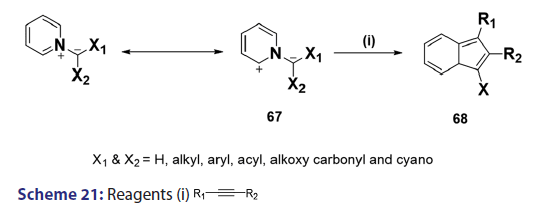
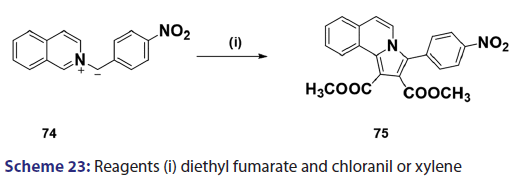
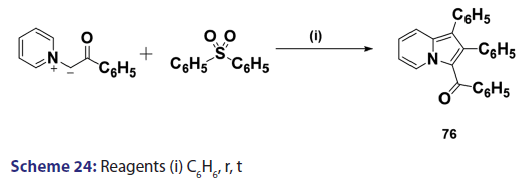
1,3?Dipolar cycloadditions are among the most widely?used reactions- in the synthesis of heterocyclic compounds, particularly 5?membered- ring compounds. It has been established that pyridinium ylides (67), even in the absence of a dehydrogenation catalyst, combine with- dimethyl acetylenedicarboxylate, diethyl acetylene dicarboxylate- methyl propynoate, ethyl propynoate, and dicyano acetylene, to- mention a few, to form the indolizines (68) [Scheme 21].[54?59] However,- when the dipolarophile is an ethnic compound, the reaction often does- not yield the indolizine directly but the tetrahydro indolizines (69)- and the dihydro indolizines (70) and (71) are isolated, subsequent- dehydrogenation in the presence of a catalyst such as palladium- on carbon, chloranil, or l, 4?benzoquinone affords the respective- indolizines. The relative rates of addition of the ylides (67) to the- dipolarophile were found to be dependent on the substituents X1- and X2 [Scheme 21]. A desirable feature of indolizine synthesis by- 1,3?dipolar cycloaddition is that the procedures are generally simple- and require only two steps. In 1961, Boekelheide and Fahrenholtz[54]- used this approach for the first time, to synthesize the indolizine (73)- from 1?phenacylpyridinium methylide (72) under dehydrogenating- conditions [Scheme 22]. Not long after, Huisgen et al.[60] used this- synthetic principle, to develop indolizine derivatives (75) from the- reaction of the azomethine (74) and dimethyl fumarate (DMF)- [Scheme 23]. Of particular interest, there was a paper published in- 1973;[37] they found that diphenylthiirene?S, S?dioxide behaved like- acetylenic compounds and reacted with pyridinium methylid to afford,- in 34% yield, the indolizine (76) [Scheme 24].
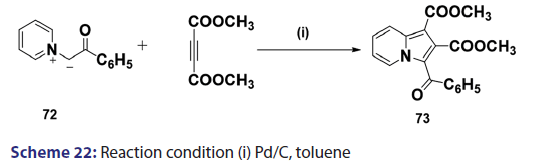
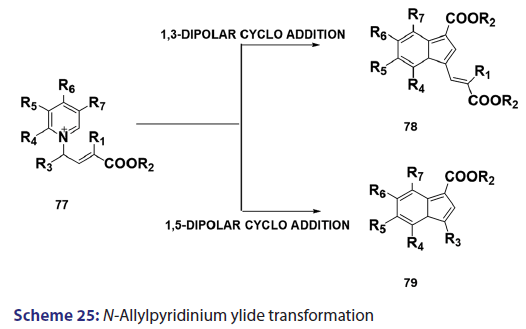
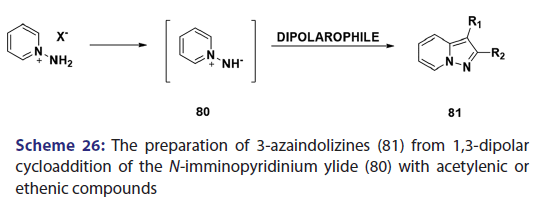
In the same year, research done on N?allylpyridinium ylides (77)- by two Japanese research groups [61?63] established that, in some- cases, N?allylpyridinium ylides behaves not only as 1,3?dipoles- which may cyclo add to another N?allylpyridinium ylide to afford the indolizine (78) but also as 1,5?dipoles, which undergo ring- closure to give the indolizines (79) [Scheme 25]. The preparation- of 3?azaindolizines (81) can be readily achieved by 1,3?dipolar- cycloaddition of the N?imminopyridinium ylide (80) with acetylenic or- ethenic compounds [Scheme 26].
Synthesis of indolizines by 1,5?dipolar cyclization
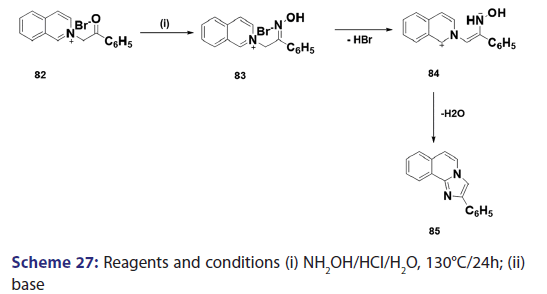
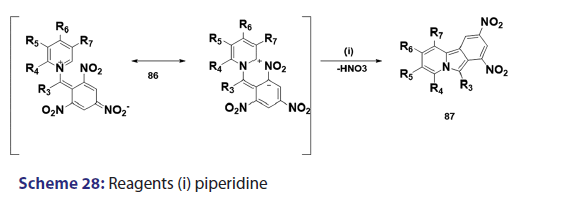
1,5?Dipolar cyclization is one of the most versatile routes to- heterocyclic molecules. Because of its utility and inherently broad- scope, this synthetic approach is of considerable importance and- Huisgen et al. studied this approach in detail. In 1962, Krohnke and- Zecher successfully extended this approach to the synthesis of the aza- indolizine (85) from phenacylisoquinolinium bromide (82) through the 1,5?dipolar intermediate (84) [Scheme 27]. The authors also- reported the synthesis of several benzindolizines (87) [Scheme 28] by- simply treating the N?picrylmethylcycioimmonium ylides (86) with- a base.
Structure and Physical and Chemical Properties of Indolizines
Structure of indolizines
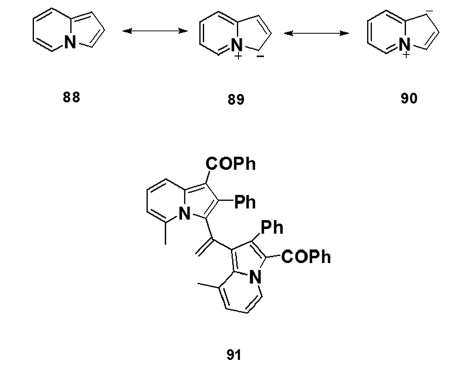
Following an early argument that any resonance stabilization in- indolizines is simply due to the presence of the pyrrole ring, the- parent indolizine was first considered to be the best represented- structure (88). However, the resonance energy (RE) calculated- for indolizine was found to be 0.29 kcal/mol, which is larger- than the total RE of pyrrole (0.23 kcal/mol). Furthermore, NMR- studies have conclusively established delocalization throughout- both rings. Therefore, indolizine is now considered to be best- represented by a resonance hybrid to which the canonical- structures (88), (89), and (90) contribute. X?ray analysis has shown- the crystal structure of the bis?indolizine (91) to be nearly planar- and the observed bond lengths to correlate well with the Huckel- molecular orbital (HMO) bond orders. HMO calculations give the- decreasing order of the electron density as: 3> 1 > >8a > 5 > 2 > 7> 6- on the ring carbons.[64]
Physical properties of indolizines
The p arent i ndolizine (2) a nd i ts a lkyl d erivatives a re e ither l ow?melting- solids or high?boiling liquids. It is sensitive to air, light and volatile in- steam. However, when a phenyl group is attached at the 2nd or 5th position,- the indolizines are found to be stable solids and nonvolatile in steam. Most- indolizines are highly fluorescent and show feeble basic properties.[1]
Chemical properties of indolizines
Reference has already been made to the structure of indolizines and,- in particular, to its delocalized orbitals. Indolizines readily undergo- electrophilic substitution and show resistance to nucleophilic attack.- In their chemical reactivity, indolizines resemble pyrroles, indoles, and- isoindoles. The following section deals with reactions of indolizines with- electrophiles, oxidation, reduction, and other miscellaneous reactions.
Reactions with electrophiles
Electrophilic substitution in indolizines occurs preferentially at the- 3?position and then at the 1?position but sometimes at both 3 and 1- positions simultaneously.[65] The e n hanced s u sceptibility t o e l ectrophilic- attack at C?3 and C?1 is consistent with the MO calculations, which indicate- C?3 to be the most reactive site for electrophilic attack, followed by C?1.
Protonation
Fraser [66] studied the protonation of indolizines using NMR spectroscopy.- Protonation of indolizines occurs preferentially at position 3. In- three?substituted indolizines, the site of protonation was found to be- dependent on the nature of the C?3 substituent as well as the substituent’s- at positions 1, 2, and 5. Indolizines which have the same substituents- at position 1 and 3 are exclusively protonated at position 3. Similarly,- protonation of 3,5?disubstituted indolizines occurs preferentially at- position 3. Fraser argued that in the 3,5?disubstituted indolizines,- intramolecular overcrowding encourages protonation at site 3. In general,- protonation of the 3?substituted indolizines affords a m i xture o f t h e- 3H? and 1H?cations [Table l]. It was found that the ratio of the 3H: 1H- cations could be increased by introducing substituents at position 2. As- indicated above, the resulting steric interaction between the C?3 and C?2- substituents is relieved by protonation at C?3. The r elief i n s teric s train- may be attributed to the consequent change in the hybridization state of- C?3. Protonation of most of the aza indolizines is found to occur at the- nonbridgehead nitrogen. Surprisingly, protonation of the 5?azaindolizines- occurs preferentially at the 3?position, followed by the 1?position.[32]
| Compound | 3H-cation | 1H-cation |
|---|---|---|
| 3-methylindolizine | 21 | 79 |
| 2,3-dimethylindolizine | 41 | 59 |
| 3-methyl-2-phenylindolizine | 72 | 28 |
| 3-methyl-2-methylindolizine | 78 | 22 |
| 1,2,3-trimethylindolizine | 100 | |
| 1,3-dimethyl-2-phenylindolizine | 100 | |
| 3,5-dimethylindolizine | 100 |
Table 1: Percentage composition of protonated 3-substituted indolizines in trifluoroacetic acid
Nitration
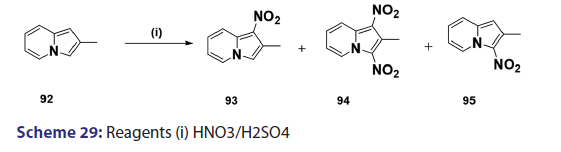
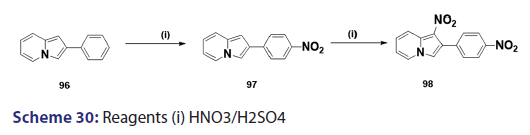
Nitration of the indolizine nucleus often results in oxidation of the- substrate with little evidence of nitration. Successful nitration the indolizine nucleus was achieved for the first time in 1946, by- Borrows et al.,[67] who showed that while the action of nitric acid on- 2?methyl and 2?phenylindolizines at moderate temperatures resulted- mainly in oxidation, rapid reaction at higher temperatures gave- the respective 1,3?dinitro indolizines in low yields. Furthermore,- nitration of 2?methylindolizine (92) in sulfuric acid has been shown- to afford the l?nitro?2?methylindolizine (93) as the main product,- accompanied by small quantities of the 3?nitro derivative (95) and- the 1,3?dinitro derivative (94) [Scheme 29]. A similar treatment of- the 2?phenylindolizine (96) resulted in the phenyl ring being attacked- first, giving the 2?p?nitrophenylindolizine (97) [Scheme 30]. This- reaction shows that the indolizine system is closely allied to pyrrole- in its properties, for similar treatment of N?phenylpyrrole yields- N?p?nitrophenylpyrrole. Compound (97) on further nitration affords- 1?nitro?2?p?nitrophenylindolizine (98) [Scheme 30]. Nitration of- 3?acetyl?2?methylindolizine proceeds readily in concentrated sulfuric- acid to yield the 1?nitroderivative along with a small quantity of- the 1,3?dinitro compounds. On the other hand, nitration of the- 3?acetyl?2?phenylindolizine under similar conditions affords a mixture- of nitrated products.
Nitrosation
Direct nitrosation of the indolizine nucleus was first achieved by Konde- and Nischizawa, in 1937, when they treated 3?acetyl?2?methylindolizine- with nitrous acid to give the l?nitroso derivative. In indolizines,- which have their 3?position unsubstituted, nitrosation takes place at- the 3?position. The preferential nitrosation at position 3 is in marked- contrast to the preferential nitration at position 1, as discussed above.
Halogenation
Not much work has been done on the halogenation of indolizines.- Attempts to prepare stable bromo derivatives have not always been- successful. However, preparation of stable iodo derivatives has proved- possible. For example, iodination of 3?acetylindolizine in alcohol- proceeds readily to give the 1,3?diiodo derivative and in the presence of- sodium acetate, the 1?iodo derivative.
Acylation
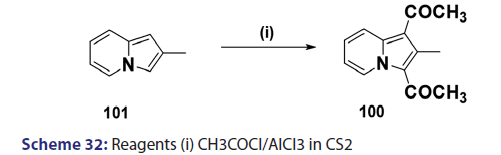
Acylation of indolizines takes place preferentially at position 3 and less- readily at position 1. Indolizines can be acylated simply by treating with acid- chlorides, anhydrides, and even esters. One of the most convenient methods- for acylating the indolizine nucleus is by heating the indolizine with acid- anhydrides in the presence of sodium salt of the corresponding acid. In 1912,- Scholtz [1] synthesized for the first time, the 3?acetyl derivative of the indolizine- and the 7?methylindolizine using this approach. Tschitschibabin [1] in 1929- and Borrows et al. in 1946 [40] extended this approach to the preparation of- 3?acetyl derivatives of 2?methyl and 2?phenylindolizine and the 3?benzoyl- derivative of 2?phenylindolizine. The monoacetyl indolizines, on further- treatment with acetic anhydride at higher temperatures, were found to yield- the respective 1,3?diacetylindolizines. In 1940, Ochai, a Japanese researcher,- reported the preparation of 1,3?diacetyl?2?methylindolizine (100) from a Friedel–Crafts reaction in CCl4 on 3?acetyl?2?methylindolizines (99)- using acetyl chloride and a large excess of aluminum chloride as catalyst- [Scheme 31]. Acylation of 2?methylindolizine (101) under similar conditions- but using carbon disulfide as solvent afforded the diacetyl derivative (100)- in very low yield [Scheme 32]. With 2?phenylindolizine, however,- Friedel?Crafts acylation proceeds readily in carbon disulfide to give a- mixture of 1,3?diacetyl?2?phenylindolizine and 2?p?acetylphenylindolizine.- It was found that acetyl chloride and bromide failed to react in the absence- of a catalyst. This is in marked contrast to the facile mono benzoylation- of indolizine when treated with benzoyl chloride even in the absence of a- catalyst.
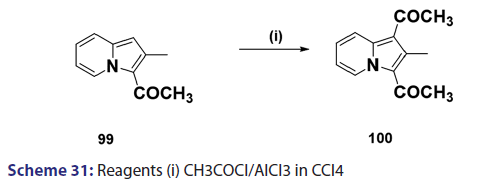
Reactions with diazonium electrophiles
The preparation of azo derivatives of indolizines can be easily achieved- using arene diazonium ion as an electrophile. Diazo coupling occurs- normally at position 3, however if this position is already occupied,- position 1 is attacked resulting in the formation of the 1?azo derivatives.- In 1913, Scholtz and Fraude achieved the first synthesis of a 3?azo- derivative of indolizine. Similar treatment of 3?acetyl?2?methylindolizine- by Kondo et al.[1] in 1936, yielded the 1?phenylazo derivative.
Oxidation reactions
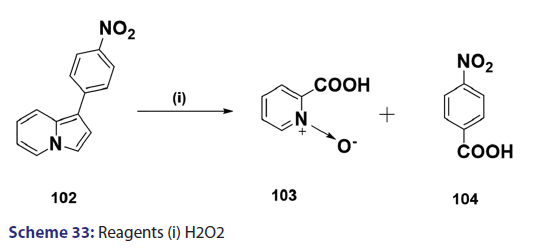
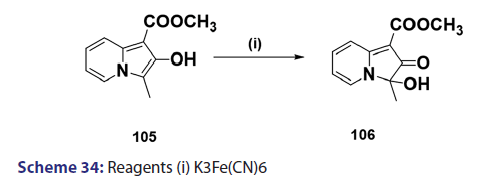
Indolizines undergo oxidation very easily. Ring fission is a rather- common phenomenon observed in oxidation of indolizines. In the past,- this reaction was used for structural elucidation.[68] A typical example is- H2O2?induced oxidation outlined in Scheme 33. However, some cases of- oxidation where ring fission does not occur have been reported. A classic- example is the potassium ferricyanide oxidation of the indolizine (104),- which afforded compound (105) as the oxidation product [Scheme 34].[69]
Reduction reactions
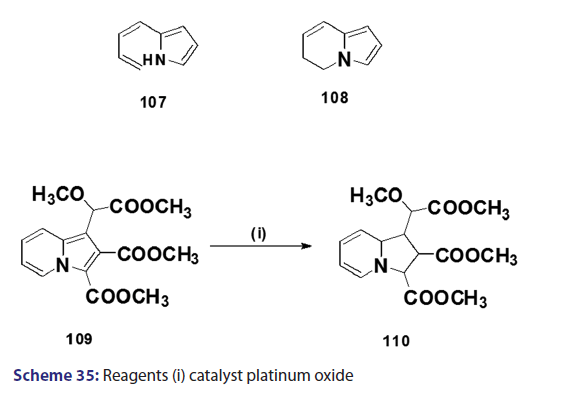
Reduction of the indolizine nucleus was first achieved in 1912 by- Scholtz,[1] when he treated the parent indolizine (2) with sodium and alcohol. Scholtz presumed the structure of the reduction- product to be the ring opened system (107). In 1946, Borrows and- Holland [1] proposed the product of the sodium?alcohol reduction- as, in fact, the dihydro derivative (108). Several reports on the- complete hydrogenation of the six?membered ring of the indolizine- system have been published. Although the above results indicate- that the six?membered ring of the indolizine nucleus is more- susceptible to hydrogenation than the five?membered ring, Diels- and Meyer reported hydrogenation of the five?membered ring- in the reduction of dimethyl 1?(methoxy carbomethoxy methyl)- indolizine?2,3?dicarboxylate (109) in the presence of platinum- oxide, the isolated product being the tetrahydro compound (110)- [Scheme 35]. Under more drastic conditions, e.g., in the presence of- Raney nickel at high temperature and pressure, complete reduction of- the indolizine nucleus has been reported.[1]
Reactions with nucleophiles and bases
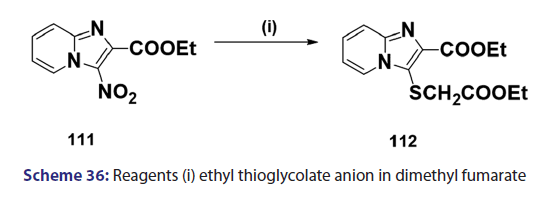
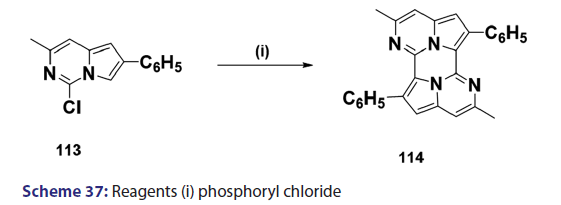
Indolizines and aza indolizines with electron withdrawing- groups undergo nucleophilic attack. Thus, treatment of 8?nitro- indolizines with secondary amines and oxygen was found to give- 5?amino?8?nitro indolizines. Similarly, 1?azaindolizine (111),- on treatment with ethyl thioglycolate anion in DMF, afforded- the thio derivative (112) [Scheme 36]; 6?azaindolizine (113), on- treatment with phosphoryl chloride, gave the bis?nitrogen bridged- annulene (114) [Scheme 37].[70]
Industrial applications of indolizines
Indolizines find use as fabric brightening agents and as photographic- sensitizers.[71] Some indolizines have been successfully used as dyes- which show great resistance to light and heat.[72]
Pharmacological properties
In 1960, James M. Price [73,74] of Wisconsin Medical School suggested the- possibility of preparing pharmacologically active indolizine derivatives by- replacing the indole ring of biologically active indoles with the indolizine- ring system. The striking structural similarity between the indole and- indolizine nucleus prompted this speculation. Most of the naturally- occurring pharmacologically active indoles such as reserpine (115), 57- lysergic acid diethylamide (116), and psilocin (116a) have the indole- nitrogen and the extra indole nitrogen separated by four carbons.[75]
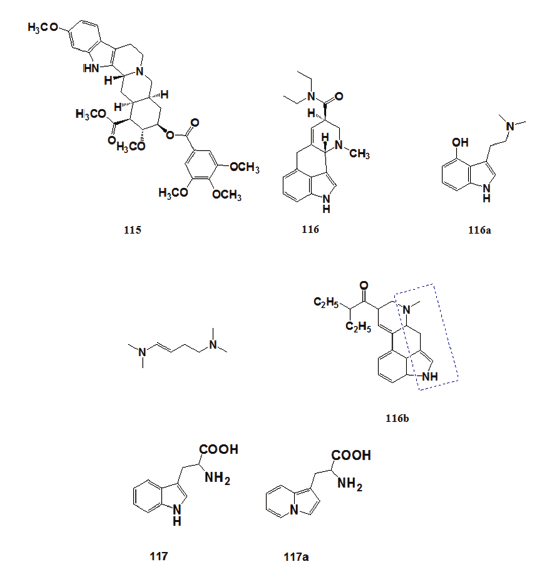
Research on these compounds suggested that their biological activity- is partly dependent on the separation between the indole nitrogen and- the extra indole nitrogen (116b). This hypothesis provided the lead for- developing indolizine analog of biologically active indoles. In 1961, Carbon- and Brehm[76,77] prepared β?(1?indolizyl) alanine (117a) as an analog of- tryptophan (117), an essential amino acid present in relatively small amounts- in proteins. Tryptophan is the precursor of several physiologically important- metabolites but is completely destroyed during acid hydrolysis of proteins.- Compound (117) is considered to be a potential tryptophan antimetabolite.
Central nervous system?depressant activity
By the late sixties, medicinal chemists in several laboratories had recognized- the importance of preparing aminoalkyl indolizines for pharmacological- studies. 1?(Diethylaminomethyl)?3?methyl?2?phenylindolizine (118),- prepared in 1966, showed depressant activity on the central nervous- system (CNS).[78] The LD50 was found to be in the range 70–100 mg/kg.- A year later, 2?phenylindolizines (119a?m) and their derivatives were- synthesized and screened for their effects on the CNS in mice and in some- cases in cats [Table 2]. Many of these compounds (e.g., 119b, d, g, h, l)- were stimulants at low doses, depressants at higher doses, and lethal at- even higher doses. Although compound (119c), at doses of 30–100 mg/kg,- produces slight CNS depression, it was found to be nonlethal at dosages- as high as 1000 mg/kg, while compound (l19e) led to a loss of aggression- in rats. Compound (119f), however, showed locomotor depression at low- doses, which become worse with increased dosage.
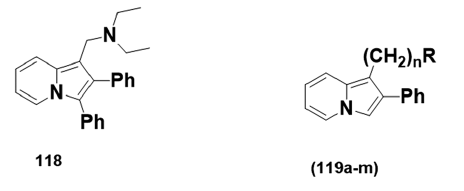
| N | R | |
|---|---|---|
| a | 1 | NCH3(COOC2H5) |
| b | 1 | N (CH3)2 |
| c | 2 | NHCOOC2H5 |
| d | 2 | NHCH3 |
| e | 2 | NCH3(COOC2H5) |
| f | 2 | N(CH3)2 |
| g | 2 | CON(CH3)2 |
| h | 3 | N(CH3)2 |
| i | 2 | N (CH3)3BR |
| j | 2 | N (CH3)3I |
| k | 2 | NHCOOC2H5 |
| l | 2 | NHCH3HCL |
| m | 2 | N (CH3)2HCL |
Table 2: Compounds (119a–m) tested for central nervous systemdepression activity.
Analgesic and anti?inflammatory activities
In 1971, certain indolizine?1?acetic acids (120) that exhibited- analgesic and anti?inflammatory activities [79?81] were developed. The- resemblance between the structure and pharmacological properties of- these indolizines to indomethacin (121), a potent anti?inflammatory- agent (introduced in 1963), suggested that the elementary constitution- necessary for anti?inflammatory action was not destroyed by the shift of- the “indole nitrogen” (in indomethacin) to the bridgehead position.[82]- This encouraged the development of a range of indolizine analogs with- anti?inflammatory properties.

Anticancer activity
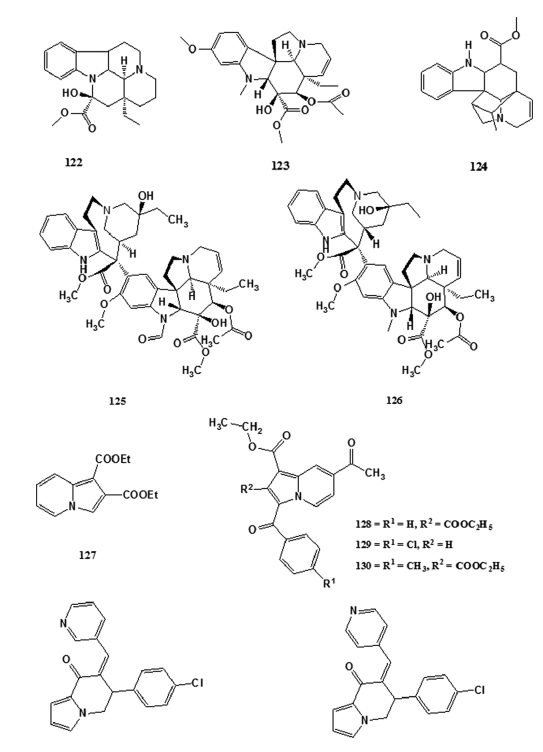
The occurrence of the indolizine ring system (2) in natural products- is not very common. However, alkaloids of the Vinca group, such- as vincamine (122), vindoline (123), and vindolinine (124), contain- several ring systems including the indolizine system.[83] Certain- indolizines were also screened for anti?neoplastic activity based on an extension of the rationale that some carcinolytic Vinca alkaloids,- such as vincristine (125) and vinblastine (126) (used in cancer- chemotherapy). However, sporadic attempts in developing carcinolytic- indolizines met with little and only one indolizine derivative, viz.,- diethyl indolizine?l, 2?dicarboxylate (127) showed significant- anticancer activity.[84,85] Sandeep et al. reported dose?dependent- anticancer activity of indolizine analogs (128–130) against human- cervix cancer cell line SiHa at 10, 20, 40, and 80 µg/mL.[86] Inhibitors- of the cytochrome P450 aromatase are therapeutic agents for the- treatment of estrogen?dependent diseases such as breast cancer.[87]- Several inhibitors of aromatase have been reported and are either- clinically available or under clinical evaluation; however, many of- these inhibitors are not as potent as expected in vivo or have terrible- side effects.[87] Thus, the synthesis and development of more powerful- and specific aromatase inhibitors are of great importance for the- treatment of breast cancers.[87] Molecular modeling studies carried out- by Sonnet et al. led to the design of a novel hypothetical aromatase- inhibitor.[87] The group synthesized an array of indolizine compounds- based on the indolizine pharmacophore and were tested for aromatase- inhibition in 30 human placental microsomes. The in vitro biological- evaluation of these compounds led to the identification of two new- and potent nonsteroidal aromatase inhibitors MR 20494 (131) and- MR 20492 (132) which are undergoing clinical development.
Antibacterial activity
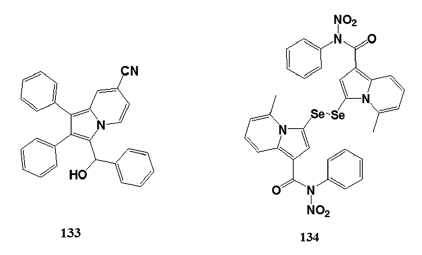
There are approximately 1.6 million deaths annually worldwide- due to tuberculosis (TB). Current treatments for TB must be- taken for 6–9 months and due to the lengthy period many- patients stop taking the drugs leading to the growing problem of multidrug?resistant strains of TB.[88] The increased resistance- of the microorganism against antibacterial compounds- calls for research and development into producing novel- TB agents which are effective against the drug?resistant TB- strains.[88] Gundersen et al. began screening indolizine derivatives- which had been previously studied for their antioxidant properties- and came across the antimycobacterial properties of (±)?1?(hydroxy- phenylmethyl)?2,3?diphenylindolizine?7?carbonitrile (133). The- indolizine compound was screened against M. tuberculosis H37Rv- and showed excellent results in vitro and with continued development- may prove to be a potent and selective antimycobacterial agent.
Antioxidant activity
Narajji et al.[89] synthesized series of indolizine compounds and- reported 3,3’?diselanediylbis (5?methyl?N?nitro?N?phenyl?indolizine?- 1?carboxamide) (134) for antioxidant activity at IC50 of- 4.11 ± 0.05 mmol L-1.
Larvicidal activity
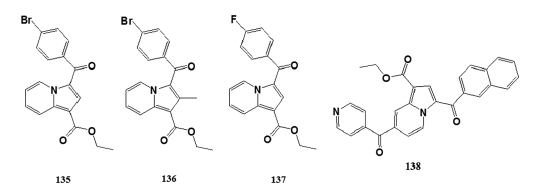
Sandeep et al.[90] reported greener synthesis of a series of novel indolizine- analogs by the cyclization of aromatic cycloimmoniumylides with electron- deficient alkynes in the presence of water as the base and solvent at 80°C.- Characterized title compounds were evaluated for larvicidal activity- against Anopheles arabiensis by the standard WHO larvicidal assay- using Temephos as standard at 4 µg/mL. Title compounds (135), (136),- and (137) exhibited promising larvicidal activity at 93%, 81%, and 95%,- respectively.
Anti?HIV activity
Huang et al.[91] reported compound (138) exhibited promising- anti?HIV?1 activity, at IC50 value of 11 µM. This information provides- new information to develop highly potent small?molecule HIV?1 virion- infectivity factor inhibitors.
Conclusion
Indolizine pharmacophore has become an important synthetic target in- the development of novel synthetic analogs with various pharmacological- properties such as CNS depressant, analgesic and anti?inflammatory,- anticancer, antibacterial, antioxidant, larvicidal, and anti?HIV.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Borrows ET, Holland DO. The chemistry of the pyrrocolines and the octahydropyrrocolines. Chem Rev 1948;42:611–43.
- Kalpana G, Komal G, Gaurav K. A pharmacognostical review on Elaeocarpus sphaericus. Int J Pharm Pharm Sci 2013;5:3–8.
- Michael JP. Indolizidine and quinolizidine alkaloids. Nat Prod Rep 1997;14:21–41.
- Takahata H, Momose T. Simple indolizidine alkaloids. Alkaloids Chem Pharmacol 1993;44:189–256.
- Angle SR, Breitenbucher JG. Recent progress in the synthesis of piperidine and indolizidine alkaloids. In: Atta–ur–Rahman, editor. Studies in Natural Products Chemistry: Stereoselective Synthesis. Vol. 16. New York; Elsevier; 1995. p. 453–502.
- Okano T, Sakaida T, Eguchi S. Synthesis of indolizidine derivatives trifluoromethylated at bridgehead position via acyl iminium ion intermediates derived from ring–chain tautomerism of 5,5,5–trifluoro–4–oxopentanoyl arylethylamides. Heterocycles 1997;44:227–36.
- Comins DL, Zhang YM. Anionic cyclizations of chiral 2,3–dihydro–4–pyridones: A five–step, asymmetric synthesis of indolizidine 209D. J Am Chem Soc 1996;118:12248–9.
- Michael JP, Gravestock D. Synthesis of (±)–Indolizidine 209B and a New 209B diastereoisomer. Synlett 1996;10:981–2.
- Giang Vo T, Célérier JP, Lhommet G. Enantioselective synthesis of (−)–indolizidine 239AB [(3R,5S,8aR)–3–butyl–5– (3–hydroxypropyl)–octahydroindolizine]. Tetrahedron Asymmetry 1996;7:2211–2.
- Chuen C, Cocker JD, Davies HG, Gore A, Green RH. Indolizidine derivatives as potential substance P antagonists. Bioorg Med Chem Lett 1996;6:161–4.
- Li Y, Marks TJ. Coupled organolanthanide–catalyzed C−N/C−C bond formation processes. Efficient regiospecific assembly of pyrrolizidine and indolizidine skeletons in a single catalytic reaction. J Am Chem Soc 1996;118:707–8.
- Takahata H, Bandoh H, Momose T. An asymmetric synthesis of the ant venom alkaloid (3S,5S,8aR)–3–butyl–5–(4–pentenyl) indolizidine via the sharpless asymmetric dihydroxylation. Heterocycles 1996;42:39–42.
- Muraoka O, Zheng BZ, Okumura K, Tabata E, Tanabe G, Kubo M. Tandem beckmann and huisgen–white rearrangement of the 9–azabicyclo[3.3.1]nonan–3–one system. Part 2.1 The second mode of the rearrangement leading to 6–(prop–1–enyl) piperidin–2–ylacetic acid, a versatile intermediate for the syntheses of piperidine alkaloids (+)–pinidine and (+)–monomorine I. J Chem Soc Perkin Trans 1 1997;2:113–20.
- Lee E, Kang TS, Chung CK. Radical cyclization of b–aminoacrylates: Expedient synthesis of (+)– monomorine 1 and (+)–indolizidine. Bull Korean Chem Soc 1996;195B: 212–4.
- Solladié G, Chu GH. Total synthesis of (+)–Indolizidine 195 B and (+)–monomorine. Tetrahedron Letters 1996;37:111–4.
- Myers CW, Daly JW. Dart–poison frogs. Sci Am 1983;248:120–33.
- Maerki F, Witkop B. The venom of the colombian arrow poison frog phyllobates bicolor. Experientia 1963;19:329–38.
- Daly JW, Garraffo HM, Spande TF. Alkaloids from amphibians. In: Cordell GA, editor. The Alkaloids. Vol. 43. San Diego: Academic Press; 1993. p. 185–288.
- Daly JW, Garaffo HM, Spande TF. In: Pelletier SW, editor. Alkaloids: Chemical and Biological Perspectives. Vol. 13. Oxford: Elsevier; 1999. p. 1.
- Daly JW, Secunda SI, Garraffo HM, Spande TF, Wisnieski A, Cover JF Jr. An uptake system for dietary alkaloids in poison frogs (Dendrobatidae). Toxicon 1994;32:657–63.
- Edwards MW, Daly JW, Myers CW. Alkaloids from a Panamanian poison frog, Dendrobates speciosus: Identification of pumiliotoxin–A and allo–pumiliotoxin class alkaloids, 3,5–disubstituted indolizidines, 5–substituted 8–methylindolizidines, and a 2–methyl–6–nonyl–4–hydroxypiperidine. J Nat Prod 1988;51:1188–97.
- Goldstein SW, Overman LE, Rabinowitz MH. The first enantioselective total syntheses of the allopumiliotoxin A alkaloids 267A and 339B. J Org Chem 1992;57:1179–90.
- Ni Y, Zhao G, Ding Y. A novel approach to the enantioselective formal synthesis of pumiliotoxin 251D. J Chem Soc Perkin Trans 1 2000:3264–6.
- Daly JW, Tokuyama T, Fujiwara T, Highet RJ, Karle IL. A new class of indolizidine alkaloids from the poison frog, Dendrobates tricolor. X–ray analysis of 8–hydroxy–8– methyl–6–(2’–methylhexylidene)–1–azabicyclo[4.3.0]nonane. J Am Chem Soc 1980;102:830–6.
- Tokuyama T, Nishimori N, Shimada A, Edwards MW, Daly JW. New classes of amidine, indolizidine and quinolizidine alkaloids from a poison–frog, dendrobates pumilio (dendrobatidae). Tetrahedron 1987;43:643–52.
- Tokuyama T, Tsujita T, Garraffo HM, Spande TF, Daly JW. Alkaloids from dendrobatid poison frogs: Further pumiliotoxins and allopumiliotoxins and a reassignment of the keto function in pumiliotoxin 307F. Tetrahedron 1991;47:5415–24.
- Kibayashi C, Aoyagi S, Wang TC, Saito K, Daly JW, Spande TF. Determination of absolute stereochemistry and an alternative synthesis of homopumiliotoxin 223G: Identification on chiral GC columns with the natural alkaloid. J Nat Prod 2000;63:1157–9.
- Gusovsky F, Padgett WL, Creveling CR, Daly JW. Interaction of pumiliotoxin B with an “alkaloid–binding domain” on the voltage–dependent sodium channel. Mol Pharmacol 1992;42:1104–8.
- Daly JW, Nishizawa Y, Padgett WL, Tokuyama T, Smith AL, Holmes AB, et al. 5,8–disubstituted indolizidines: A new class of noncompetitive blockers for nicotinic receptor–channels. Neurochem Res 1991;16:1213–8.
- Daly JW, McNeal ET, Overman LE, Ellison DH. A new class of cardiotonic agents: Structure–activity correlations for natural and synthetic analogue of the alkaloid A new class of A new class of cardiotonic agents: Structure–activity correlations for natural and synthetic analogue of the alkaloid pumiliotoxin B (8–hydroxy–8–methyl–6–alkylidene–1–azabicyclo[4.3.0] nonanes). J Med Chem 1985;28:482–6.
- Daly JW, McNeal E, Gusovsky F, Ito F, Overman LE. Pumiliotoxin alkaloids: Relationship of cardiotonic activity to sodium channel activity and phosphatidylinositol turnover. J Med Chem 1988;31:477–80.
- Daly JW, Gusovsky F, McNeal ET, Secunda S, Bell M, Creveling CR, et al. Pumiliotoxin alkaloids: A new class of sodium channel agents. Biochem Pharmacol 1990;40:315–26.
- Larry EO, Leslie AR, Jeffery Z. First total synthesis of (+)–allopumiliotoxin 339A. A practical entry to dendrobatid alkaloids of the allopumiliotoxin class. J Am Chem Soc 1992;114:368–9.
- Franklin AS, Overman LE. Total syntheses of pumiliotoxin A and allopumiliotoxin alkaloids. Interplay of pharmacologically active natural products and new synthetic methods and strategies. Chem Rev 1996;96:505–22.
- Trost BM, Scanlan TS. Stereoelectronic requirements of palladium(0)–catalyzed cyclization. A synthesis of allo–pumiliotoxin 339B. J Am Chem Soc 1989;111:4988–90.
- Uchida T, Matsumoto K. Methods for the construction of the indolizine nucleus*. Synthesis 1976;4:209–36.
- Hurst J, Melton T, Wibberley DG. 529. Indolizines. Part III. J Chem Soc (Resumed) 1965;8:2948–55.
- Melton T, Taylor J, Wibberley DG. A new synthesis of indolizines and related nitrogen–bridgehead compounds. Chem Commun London 1965;8:151b–2.
- Melton T, Wibberley DG. Indolizines. Part IV. Syntheses from 2–acylmethylene–1–benzyl–1,2–dihydropyridine, phenyl–2–picolyl sulphone, and related compounds. J Chem Soc C Org 1967:983–8.
- Borrows ET, Holland DO, Kenyon J. 236. The chemistry of the pyrrocolines. Part I. 2–Methyl– and 2–phenyl–pyrrocoline. J Chem Soc (Resumed) 1946:1069–75.
- Borrows ET, Holland DO, Kenyon J. 237. The chemistry of the pyrrocolines. Part II. Nitroso–derivatives of some substituted pyrrocolines. J Chem Soc (Resumed) 1946:1075–7.
- Bragg DR, Wibberley DG. 507. Preparation of indolizines from ethyl 2–pyridylacetate. J Chem Soc (Resumed) 1962:2627–9.
- Roberts EM, Gates M, Boekelheide V. A synthesis of 5,6–benzopyrrocoline. J Org Chem 1955;20:1443–7.
- Barrett PA, Chambers KA. 63. Aminoalkyl tertiary carbinols and derived products. Part VIII. Some 1–alkyl– and 1: 2–cycloalkano–pyrrocolines. J Chem Soc (Resumed) 1958:338–49.
- Adamson DW, Barrett PA, Billinghurst JW, Jones TS. 445. Aminoalkyl tertiary carbinols and derived products. Part V. Antihistamines. The stereochemistry of cis– and trans–3–phenyl–3–pyridylallylamines. J Chem Soc (Resumed) 1957:2315–26.
- Glover EE, Vaughan KD, Bishop DC. Synthesis and quaternization of some heterocyclic mono– and disulphides. J Chem Soc Perkin Trans 1 1973:2595–9.
- Borrows ET, Holland DO. 128. The chemistry of the pyrrocolines. Part VI. The synthesis of pyrrocoline–2–carboxylic acid: A new route to pyrrocoline. J Chem Soc (Resumed) 1947:672–4.
- Boekelheide V, Windgassen RJ. Syntheses of pyrrocolines unsubstituted in the five–membered ring 1. J Am Chem Soc 1959;81:1456–9.
- Wiley R, Knabeschuh L. Pyrrocolines from the diene synthesis with some pyridine bases and dimethyl acetylenedicarboxylate. J Org Chem 1953;18:836–41.
- Acheson RM, Plunkett AO. 729. Addition reactions of heterocyclic compounds. Part XI. The constitution of some adducts from phenanthridine and dimethyl acetylenedicarboxylate. J Chem Soc (Resumed) 1962:3758–70.
- Acheson RM, Robinson DA. Addition reactions of heterocyclic compounds. Part XXXVII. Dimethyl acetylenedicarboxylate with some substituted pyridines. J Chem Soc C Org 1968:1629–33.
- Acheson RM, Bailey AS, Selby IA. Phenanthridine 5–oxides with acetylenic ester and the preparation of dibenzo [e, g] indolizine. Chem Commun London 1966:835a.
- Boekelheide V, Godfrey JC. Syntheses of 7,8–benzopyrrocoline derivatives. A novel reaction of reissert compounds. J Am Chem Soc 1953;75:3679–85.
- Boekelheide V, Fahrenholtz K. The formation of pyrrocolines by the reaction of dimethyl acetylenedicarboxylate with heterocyclic zwitterions 1. J Am Chem Soc 1961;83:458–62.
- Linn WJ, Webster OW, Benson RE. Tetracyanoethylene Oxide. I. Preparation and Reaction with nucleophiles. J Am Chem Soc 1965;87:3651–6.
- Sasaki T, Kanematsu K, Yukimoto Y, Ochiai S. Heteroaromaticity. XLIII. Orientation in the 1,3–dipolar cycloaddition reactions of heteroaromatic nitrogen methylides with dipolarophiles. J Org Chem 1971;36:813–8.
- Henrick C, Ritchie E, Taylor W. Pyridinium ylids in synthesis. III. Synthesis of indolizines. Aust J Chem 1967;20:2467–77.
- Basketter N, Plunkett AO. Some 1,3–dipolar addition reactions of isoquinolinium ylides: Formation of 2,3–dihydrobenzo[g] indolizines and benzoindolizines. J Chem Soc D Chem Commun 1971;23:1578.
- Farnum DG, Alaimo RJ, Dunston JM. Synthesis of azaindenes. The benzo[c] pyrazolo[1,2–a] cinnolinium cation, a novel heteroaromatic cation. J Org Chem 1967;32:1130–4.
- Huisgen R, Grashey R, Steingruber E. Azomethin–ylide und ihre 1.3–dipolaren cycloadditionen. Tetrahedron Lett 1963;4:1441–5.
- Sasaki T, Kanematsu K, Kakehi A, Ito GI. Molecular design by cycloaddition reactions. Part IX. Further investigation of the cycloaddition reactions of pyridinium allylides [1–(1–pyridinio) prop–2–enides] to give indolizines. J Chem Soc Perkin Trans 1 1973:2089–91.
- Sasaki T, Kanematsu K, Kakehi A, Ito G. Studies of heteroaromaticity–LXIV. Tetrahedron 1972;28:4947–58.
- Tamura Y, Sumida Y, Ikeda M. Synthesis and thermal reaction of pyridinium 3,3–diacyl–1–benzoylallylides [3,3–diacyl–1–benzoyl–1–(1–pyridinio) prop–2–enides]: Formation of indolizine derivatives. J Chem Soc Perkin Trans 1 1973:2091–5.
- Flitsch W, Katritzky AR, Rees CW. Comprehensive Heterocyclic Chemistry. Vol. 1. Pergamon, Oxford; 1984. p. 445.
- Fraser M, McKenzie S, Reid DH. Nuclear magnetic resonance. Part IV. The protonation of indolizines. J Chem Soc B Phys Org 1966:44–8.
- Fraser M. Azaindolizines. I. Protonation of 5–azaindolizine. J Org Chem 1971;36:3087–91.
- Borrows ET, Holland DO, Kenyon J. 238. The chemistry of the pyrrocolines. Part III. Nitration. J Chem Soc (Resumed) 1946:1077–83.
- Jones G, Sidgwick V, Millar IT, Springall HD. The Organic Chemistry of Nitrogen. 3 ed. Oxford: Springall Clarendon Press; 1966. p. 752.
- Bowers RJ, Brown AG. Oxidation of methyl 2–hydroxy–3–methyl– and methyl 2–hydroxy–3–phenylindolizine–1–carboxylates. J Chem Soc C Org 1970;10:1434–6.
- Flitsch W, Katritzky AR, Rees CW. Comprehensive Heterocyclic Chemistry–I. Vol. 4. Pergamon, Oxford:1984. p. 443–56.
- Bode ML, Kaye PT. Indolizine studies. Part 2. Synthesis and NMR spectroscopic analysis of 2–substituted indolizines. J Chem Soc Perkin Trans 1 1993;15:1809–13.
- Weidner CH, Wadsworth DH, Bender SL, Beltman DJ. Indolizines. 4. Dyes derived from oxoindolizinium ions and active methylene compounds. J Org Chem 1989;54:3660–4.
- Flitsch W, Katrizky AR, Rees CW, Scriven EF. Comprehensive Heterocyclic Chemistry II. Vol. 8. Pergamon, Oxford; 1996. p. 459.
- Armarego WL. Ionization and ultraviolet spectra of indolizines. J Chem Soc (Resumed) 1964:4226–33.
- Harrell WB, Doerge RF. Mannich bases from 2–phenylindolizines 3. 1,3–bis (dialkylaminomethyl)–2–phenylindolizines. J Pharm Sci 1968;57:1989–91.
- Carbon JA, Brehm S. 1–Indolizinealanine – A possible tryptophan antimetabolite. J Org Chem 1961;26:3376–9.
- Jones G, Stanyer J. The mass spectra of indolizines. Org Mass Spectrom 1970;3:1489–98.
- Harrell WB, Doerge RF. Mannichbasesfrom 2–phenylindolizines. I. 3–Alkyl–1–dialkylaminomethyl derivatives. J Pharm Sci 1967;56:225–8.
- Black P, Heffernan M, Jackman L, Porter Q, Underwood G. Proton magnetic resonance spectra of indolizine and its methyl derivatives and aza analogue. Aust J Chem 1964;17:1128–37.
- Pugmire RJ, Smith JC, Grant DM, Stanovnik B, Tišler M, Ver?ek B. Correlation of ring nitrogen substituents with carbon–13 nuclear magnetic resonance data in azoloazines. J Heterocycl Chem 1987;24:805–9.
- Grant DM, Pugmire RJ, Robins MJ, Robins RK. Carbon–13 magnetic resonance. XX. 4–Azaindene (pyrrocoline) and related bridgehead nitrogen heterocycles. J Am Chem Soc 1971;93:1887–93.
- Kallay KR, Doerge RF. p–Substituted 1,2–diphenylindolizines as anti–inflammatory agents. J Pharm Sci 1972;61:949–51.
- De AU, Saha BP. Search for potential oral hypoglycemic agents: Synthesis and activity of 2–(N–alkylaminomethyl) indolizines. J Pharm Sci 1973;62:1897–8.
- De AU, Saha BP. Indolizines II: Search for potential oral hypoglycemic agents. J Pharm Sci 1975;64:49–5.
- Crews P, Kintner RR, Padgett HC. Localization or delocalization of nonbonded electrons in unsaturated heterocycles. J Org Chem 1973;38:4391–5.
- Sandeep C, Basavaraj P, Venugopala KN, Rashmi SK, Rashmi V, Odhav B. Efficient synthesis and characterization of ethyl 7–acetyl–2–substituted–3–(substitutedbenzoyl) indolizine–1–carboxylates for in–vitro anticancer activity. Asian J Chem 2016;28:1043–8.
- Sonnet P, Dallemagne P, Guillon J, Enguehard C, Stiebing S, Tanguy J, et al. New aromatase inhibitors. Synthesis and biological activity of aryl–substituted pyrrolizine and indolizine derivatives. Bioorg Med Chem 2000;8:945–55.
- Gundersen LL, Charnock C, Negussie AH, Rise F, Teklu S. Synthesis of indolizine derivatives with selective antibacterial activity against Mycobacterium tuberculosis. Eur J Pharm Sci 2007;30:26–35.
- Narajji C, Karvekar MD, Das AK. Synthesis and antioxidant activity of 3,3’–diselanediylbis (N, N–disubstituted indolizine–1–carboxamide) and derivatives. S Afr J Chem 2008;61:53–5.
- Sandeep C, Venugopala KN, Gleiser RM, Chetram A, Padmashali B, Kulkarni RS, et al. Greener synthesis of indolizine analogue using water as a base and solvent: Study for larvicidal activity against Anopheles arabiensis. Chem Biol Drug Des 2016;899–904.
- Huang W, Zuo T, Luo X, Jin H, Liu Z, Yang Z, et al. Indolizine derivatives as HIV–1 VIF–ElonginC interaction inhibitors. Chem Biol Drug Des 2013;81:730–41.